Spinal muskelatrofi hos børn

Spinal muskelatrofi er en alvorlig patologi, der ofte gør simple handlinger, såsom at gå, sidde, utilgængeligt for et barn. Barnet kan fratages selv en sådan naturlig mulighed som uafhængig vejrtrækning. Det er meget svært at lave forudsigelser for denne patologi, Der findes trods alt ingen særlig behandling eller profylakse, men alt afhænger af sygdommens og faktorernes form, hvilken medicin kan ikke finde en forklaring.
Hvad er det her?
Talende om spinal muskelatrofi indebærer ikke en specifik sygdom, men en hel gruppe sygdomme under den generelle forkortelse CMA. Alle er arvelige og er forbundet med degenerering af nervecellerne i rygmarven, som er ansvarlige for motorfunktioner.
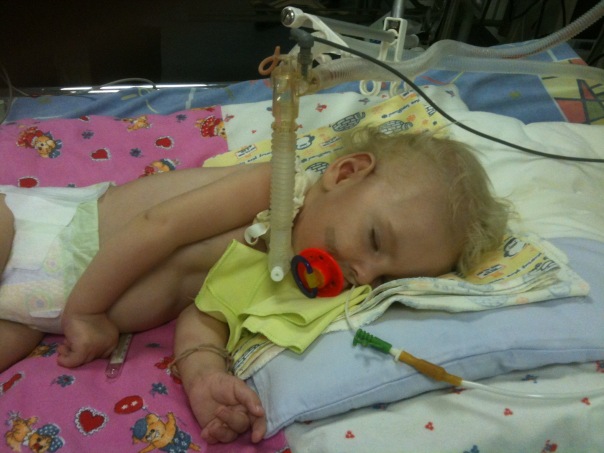
Blandt de genetiske patologier hos børn spinal muskulære atrofier optager førende steder med hensyn til frekvens af spredning. Og omkring en ud af 6 tusinde børn er født med en sådan forfærdelig diagnose. I 50% af tilfældene lever børn ikke i to år og dør. Restenes liv er handicap.

Problemet, ifølge genetikere, er meget bredere end det kan forekomme fra de givne statistikker.
Sygdommen udvikler sig på grund af mutationen af visse gener, og en af dem er SMN1, som betragtes som den vigtigste "synder" af patologi, i en modificeret form for recessivt til stede i hver halvtredsindbygger på planeten. Det betyder, at sunde forældre, der ikke engang er klar over, at de er recessive bærere af det muterede gen, kan godt have en baby med spinal muskelatrofi.
Gruppen af sygdomme blev først beskrevet i det 19. århundrede af Guido Verding, hvis navn senere blev opkaldt som en af børns sorter af AGR.
klassifikation
Den mest almindelige form for SMA hos børn er proximal. Det er repræsenteret af flere typer sygdomme, som ikke alle bliver tydelige umiddelbart efter barnets fødsel.
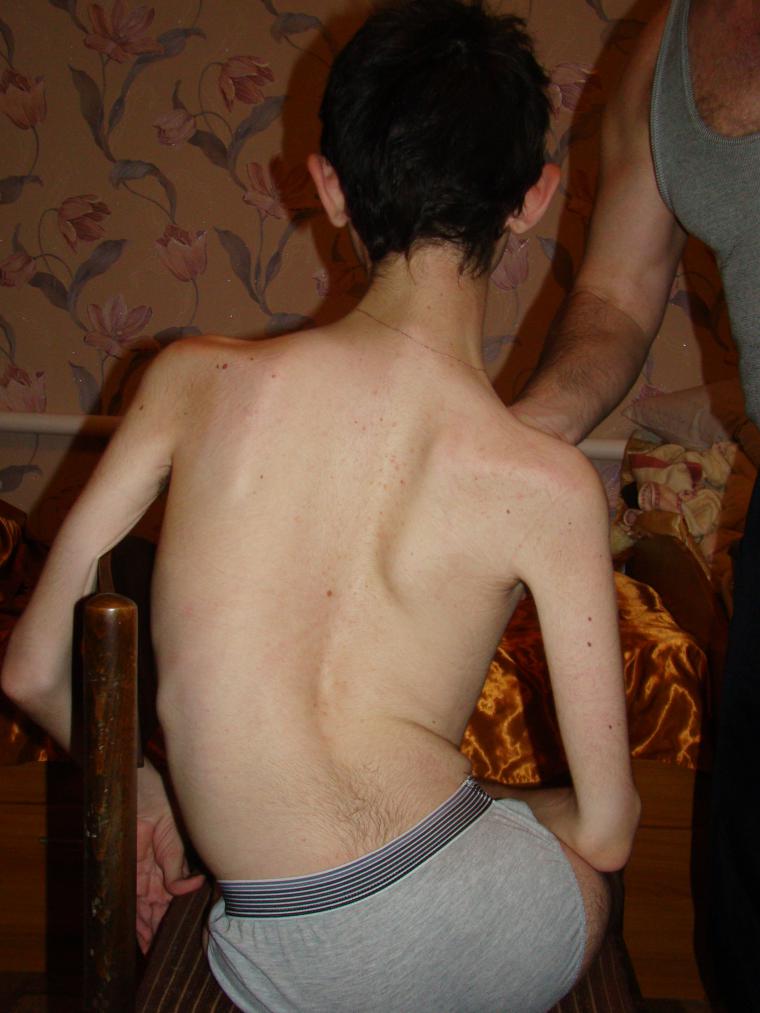
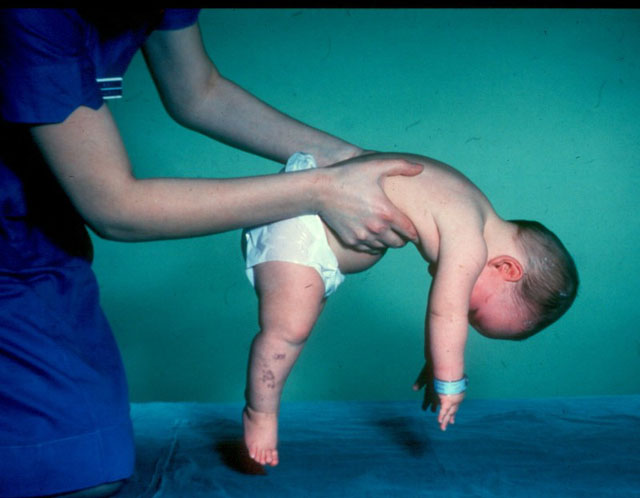
- Verding-Hoffman sygdom - Type 1 MCA, svær spædbarnssygdomsom manifesterer sig i de første seks måneder af barnets liv. Prognoser med hendes mest ugunstige, de fleste patienter dør. Et barn med type 1 SMA kan hverken stå eller sidde eller veksle selvstændigt. Mange nyfødte har nedsat sugende og synke reflekser. Ofte er der ingen mulighed for spontan vejrtrækning eller vejrtrækning er vanskelig.
- Atrofi Dubovitsa - CMA type 2, sent spædbarn. Det sker normalt mellem seks måneder og et og et halvt år senere. Barnet kan ikke gå, stå, men er i stand til at sidde, maden er ikke forstyrret, han er i stand til at sluge og sutte. Hvor længe babyen vil leve afhænger af tilstanden af åndedrætsmusklerne.
- Atrofi af Kugelberg-Welander - CMA 3 typer, infantil. Normalt fundet i en og et halvt år, normalt om to år. Prognostisk mere gunstig form. Små patienter kan stå, sidde, bevæge sig, men opleve stor svaghed og har derfor i de fleste tilfælde brug for en kørestol, uden hvilken deres normale livsvigtige aktivitet er vanskelig for dem.
- Kennedy atrofi - CMA type 4, bulbospinalnaya. Normalt betragtes som en voksen form, men lejlighedsvis opdaget hos børn efter 15 år. Indflydelse af liv påvirkes sjældent, svækkelse af muskler opstår langsomt, gradvist, en person, der førte et normalt liv og betragtede sig sundt, bliver i sidste ende deaktiveret og mister evnen til at bevæge sig selvstændigt.


Mere eller mindre kendt førstehånds atrofi af Duchenne og atrofi af Vulpian er SMA "voksne", den første er normalt detekteret efter 18 år og den anden efter 20 år.
Hos børn registreres ikke kun isolerede former for SMA, når der ikke er noget, der undgår muskeldystrofi, men også kombinerede former, når spinalatrofi ikke er den eneste diagnose, og barnet har andre genetiske eller medfødte problemer, såsom hjerte og vaskulære defekter, oligofreni.
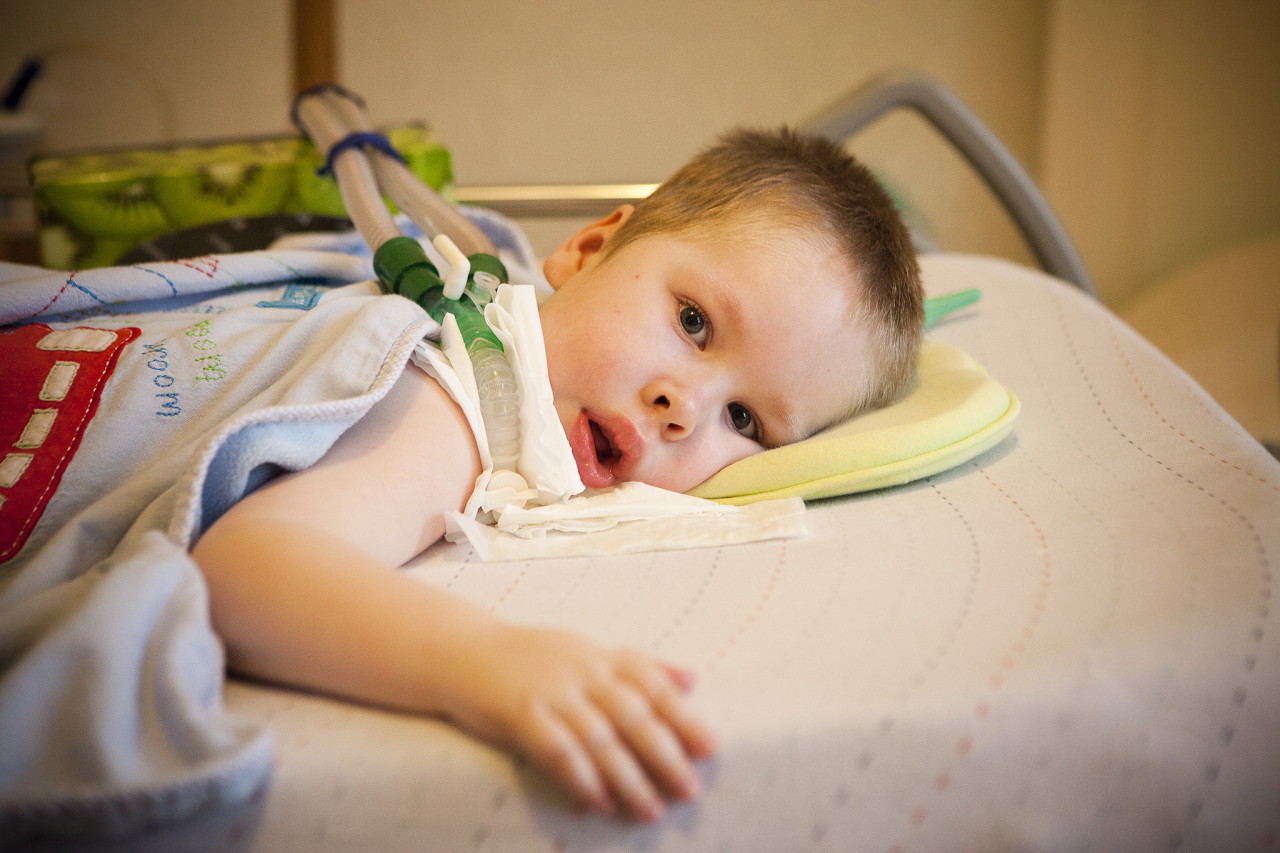
grunde
Som vi allerede nævner, taler vi om en genetisk sygdom, og derfor er årsagerne til dens forekomst et område med søgninger efter genetikere. Barnet arver et af de recessive gener på det femte kromosom (disse kan være SMN, NAIP, H4F5, BTF2p44 gener).
Sandsynligheden for at overføre et sådant gen til et afkom fra en bærer er høj - 25%. Hvis både mor og far er skjulte bærere af det muterede gen, er sandsynligheden for AGR hos et barn 50%. Det berørte abnormale gen forhindrer normal SMN-proteinproduktion, og de nerveceller, der er ansvarlige for motorerne i musklerne i rygmarven, begynder gradvist at dø. Processen med deres død fortsætter, selv efter at barnet er født.


manifestationer
Symptomer afhænger af sygdommens art. Da vi kun betragter fire typer børn, skal det bemærkes, at muskelsvaghed og muskelatrofi er karakteristiske for alle. Ellers har hver type sit eget kliniske billede og særpræg.
- CMA type 1 (Verding-Hoffman atrofi) Til rådighed til afsløring selv under graviditeten. Lægen kan mistanke om sygdommen i fosteret med meget svag omrøring. Men for at bekræfte diagnosen på tidspunktet for at bære et barn er det svært, opstår det normalt efter fødslen. Et barn med sådan atrofi kan ikke holde hovedet selv, kaste fra side til side, han sidder ikke ned. Han ligger næsten altid på ryggen, hans krop er afslappet, han løfter ikke benene, bringer dem ikke sammen, lægger ikke håndfladerne sammen. På et meget tidligt stadium kan der være store problemer for at fodre barnet, fordi han svulger det viser sig meget dårligt eller ikke virker. De fleste børn dør inden to år. Nogle formår at leve i syv eller otte år, men atrofi bliver kun værre. Normalt forekommer døden på grund af mangel på hjerte, lunger, fordøjelsesorganer.
- CMA 2 type (atrofi Dubovitsa) ved fødslen bliver det normalt ikke opdaget, fordi barnet er i stand til at trække vejret, sluge mad, og først efter seks måneder bliver fremskridt i muskelatrofi tydelig. Hvis de første symptomer opstår i en alder, hvor barnet allerede har lært at stå i krybben, så et skæreskilt på benene, kan et urimeligt fald i krummerne være et lyst tegn. Efterhånden bliver det svært at sluge. Over tid begynder barnet at have brug for en kørestol.
- CMA type 3 (amyotrofi af Kügelberg-Welander) kan dukke op i enhver alder efter 2 år. Et barn, som normalt voksede og udviklede sig, begynder at klage over svaghed, normalt i skuldrene, underarmene. Da det skrider frem, bliver det svært for ham at løbe, gå ad trapper, kneppe. Det hele afhænger af plejen - nogle bevarer evnen til at bevæge sig uafhængigt i mange år.
- CMA type 4 (Kennedy atrofi) forekommer kun hos mandlige patienter, da det anses for at være forbundet med sexchromosom X. De første tegn er svaghed i lårmuskelområdet, og kraniale nerver påvirkes gradvist. Sygdommen skrider langsomt.

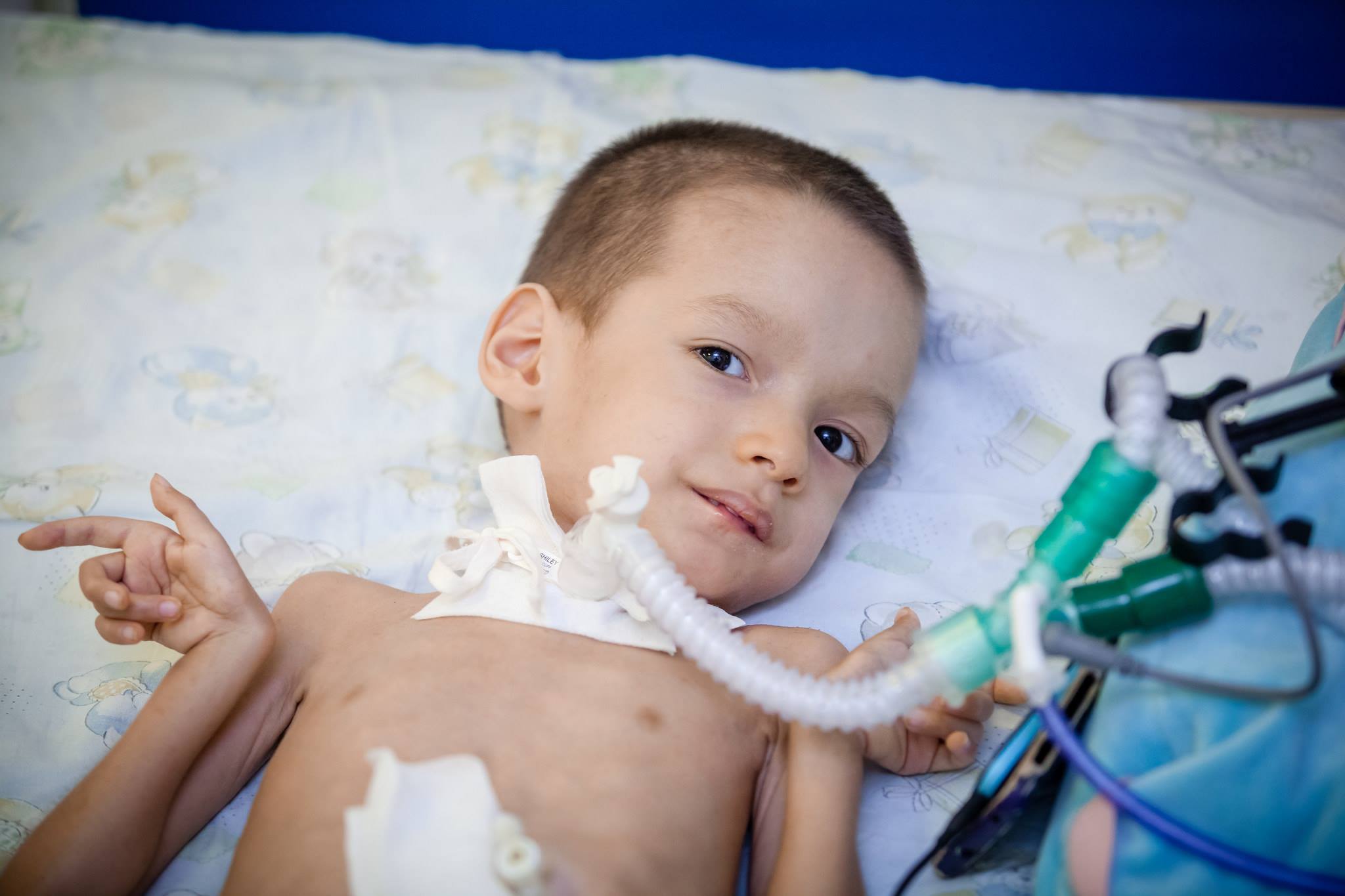
behandling
Desværre kan medicinsk viden i dag ikke tilbyde metoder og midler til behandling af SMA. Der findes ingen sådanne metoder. At opretholde kroppens funktioner og maksimere perioden, indtil barnet kan bevæge sig selv, medicin som f.eks Prozerin, Oksazil. De reducerer aktiviteten af et enzym, der er i stand til at spalte acetylcholin, som overfører en excitationspuls gennem fibrene i nervesystemet.
Anbefales også systematisk brug af stoffer, der forbedrer energiudveksling på cellulært niveau, gruppe B-vitaminer, nootropiske lægemidler samt kalium- og nicotinsyrepræparater.
Et barn med SMA højprotein kost vistMen nylige undersøgelser har vist, at diætets rolle er noget overdrevet - der er intet tegn på, at et højt indhold af protein i fødevarer i det mindste på en eller anden måde påvirker sygdommens progressionsgrad.
Men med kalorierne skal man være mere forsigtig - på grund af nedsat muskelaktivitet kan barnet hurtigt få ekstra pund.
For at hjælpe med at forlænge perioden mere eller mindre tilfredsstillende, vil livet hjælpe terapeutisk massage, UHF, elektroforese, åndedrætsøvelser til vedligeholdelse af åndedrætsmusklerne, svømning. Det anbefales at bære bærende ortopædiske rygsøjler.
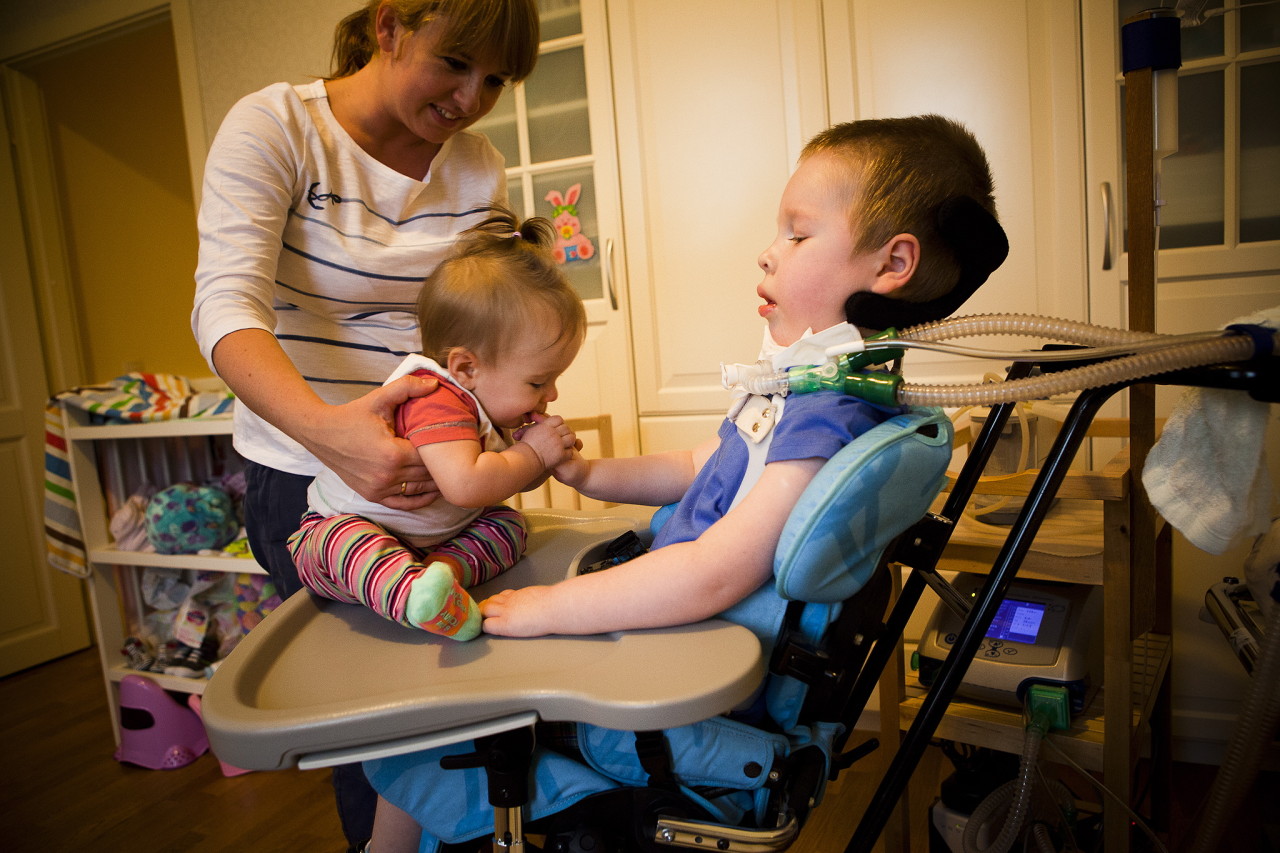
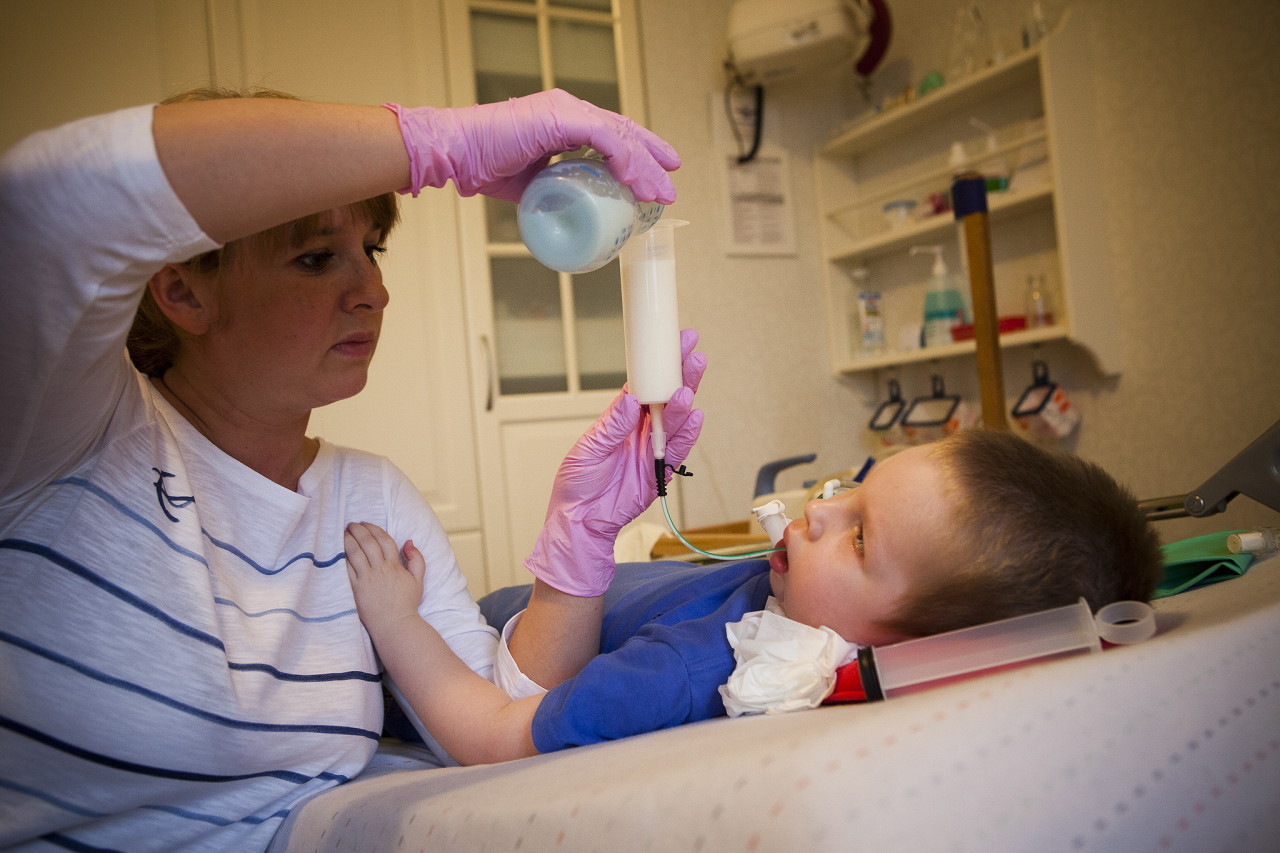

Flere oplysninger om sygdommen fortæller en specialist i videoen nedenfor.